Thomas Denecker & Claire Toffano-Nioche



Session 5
26/04/2019
Novembre
Janvier
Février
Mars
Avril
Mai
Juin
Juillet
...
26
25
22
29
26
31
28
à définir
...
Attention !!
Changement de planning
Mai
31 mai pont ?
Proposition : 24 mai ou 7 juin
Juin (changement obligatoire)
Nouvelle date : 21 juin

Docker
Vos retours
Je ne vois pas l'intérêt...



Super compliqué pour pas grand chose
Moi j'ai jamais eu de problèmes de reproductibilité entre OS
Vous avez raison ! (en partie)
Docker pas si simple?
Oui ... et non
- Mettre en place un Rstudio server : 5 minutes
- Faire son propre docker : ça demande du travail
Jamais de problèmes de reproductibilité à cause de l'OS
En effet, c'est rare mais ça peut arriver (MAJ, partage,...)
Pas énormément d'utilité?
C'est vrai, le docker ne fait rien de plus que le local
Mais Partage + Reproductibilité + Déploiement
En résumé

Session 5
I've got the power!
Reproductible et rapide
Aujourd'hui
Script automatique
Utilisation d'un cloud avec une puissance paramétrable
Que faire de plus?
Paralléliser
Sur un cluster de calcul
Programme de la session
La parallélisation
- Snakemake
- Comparaison avec le script.sh
Le cluster de calcul
- C'est quoi?
- Cluster de l'IFB / I2BC
- Singularity ↔ Docker
- Exemple sur l'IFB et l'I2BC
Parallélisation
Snakemake
Le temps de calcul
Même sur le cloud de l'IFB, c'est long...
Que faire ?
1- Améliorer les algorithmes
Prêt pour optimiser Bowtie2 ? Non...
2- Augmenter la puissance de calcul
- Comment ?
- Comment exploiter cette puissance ?
Augmenter la puissance de calcul
Solution 1 : Changer d'ordinateur $$$
Solution 2 : Augmenter la puissance de notre VM
Disque dur
RAM
CPU
20 Go
160 Go
2 Go
1
500 Go
48


CPU?
Central Processing Unit = processeur
Composant qui exécute les instructions

Intel 4004
1971
0.092 MIPS
(Million Instructions Per Second)

INTEL® CORE™ i9-7900X
2019
57411 MIPS
10 cœurs
Un cœur ?
Un processeur est composé d'un ou plusieurs cœurs
1 cœur = 1 instruction
n cœurs = n instructions

Dans notre cas
Nous avons beaucoup d'instructions donc il nous faut beaucoup de cœurs !
Pourquoi ? Pour paralléliser
Linéaire (1 cœur)

Programme 2
Programme 3
Programme 1
Programme 4
Parallélisation (n cœurs)
Plusieurs instructions simultanément
Comment paralléliser ?
Plusieurs solutions possibles : Snakemake, Nextflow,...
Installer Snakemake
conda install -c bioconda snakemakePourquoi Snakemake ?
- Système de workflow
- Lisibilité du code
- Gestion automatique des ressources
- Reproductibilité !
Le principe
Système de règles
- un nom
- des fichiers d’entrée
- des fichiers de sortie
- du code pour passer des fichiers d’entrée aux fichiers de sortie
Règle
Input
Output
Notions importantes
Snakemake doit connaître le futur
Dans l'ordre des règles :
- La première règle : les fichiers que nous voulons à la fin (cible/target)
- Ecrire les règles pour y arriver
Inputs
Cibles
Recherche de la règle précédente qui a le même output que l'input de la règle actuelle
1
2
3
4
Ordre d'exécution
Comparaison des flux
FASTQC
Bowtie2
SAMtools
DEseq2
HTSeq
6 fois
6 fois
6 fois
6 fois
1 fois
Avant
Comparaison des flux
Maintenant
F
F
F
F
F
F
B
B
B
B
B
B
S
S
S
S
S
S
H
H
H
H
H
H
C
Représentation du flux avec Snakemake
snakemake --forceall --dag | dot -Tsvg > dag_input.svg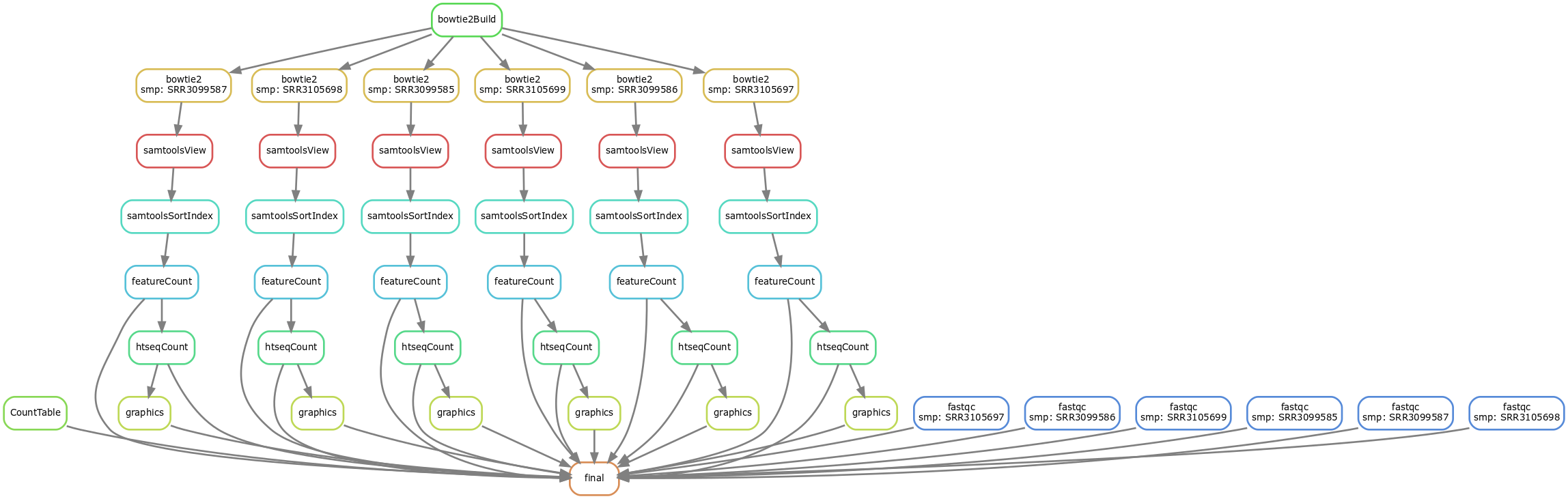
Exemple
1- Une règle pour indiquer ce que nous voulons
rule targets:
input:
"data/toto.fq.gz"
"data/titi.fq.gz"
rule gzip:
input:
"{base}"
output:
"{base}.gz"
shell:
"gzip {input}"2- Une règle pour indiquer comment y arriver
{base} : Stocke le nom des fichiers où nous nous trouvons
Ici nous compressons des fichiers
Snakefile du projet
1- Changement du dossier de travail
BASE_DIR = "/home/rstudio"
WDIR = BASE_DIR + "/Project"
workdir: WDIR
2- Déclaration de variables (GFF et GENOME)
GFF = WDIR + "/annotations/O.tauri_annotation.gff"
GENOME = WDIR + "/genome/O.tauri_genome.fna"Le snakefile est le fichier qui contient toutes les règles.
Nom du fichier : Snakefile
Snakefile du projet
glob_wildcards() : fonction qui déduit la partie manquante des noms de fichier à partir des fichiers présents dans le système
smp : la partie variable
3- Récupération de la liste des fichiers à traiter
SAMPLES, = glob_wildcards("./samples/{smp}.fastq.gz")
NB_SAMPLES = len(SAMPLES)
SAMPLES, : ne pas oublier la virgule (liste)
Snakefile du projet
expand(...) : fonction qui permet d'utiliser une variable. Ici la variable = nom des fichiers
smp : devient une variable globale
4- Déclaration des fichiers que nous souhaitons
rule final:
input:
expand("fastqc/{smp}/{smp}_fastqc.zip", smp=SAMPLES),
expand("htseq/count_{smp}.txt", smp=SAMPLES),
expand("graphics/graphics-{smp}.pdf", smp=SAMPLES),
expand("countTable.txt")Si le fichier n'est pas déclaré ici, les règles associées ne seront pas exécutées
Snakefile du projet
input : les fichiers qui ont été téléchargés pour l'analyse
output : les fichiers de sortie
message : message affiché lors de l'analyse
shell : commande lancée dans le terminal. Pour utiliser la variable : wildcards.nomVariable
5- Fastqc
rule fastqc:
input: "samples/{smp}.fastq.gz"
output: "fastqc/{smp}/{smp}_fastqc.zip"
message: """--- Quality check of raw data with Fastqc."""
shell: """
fastqc {input} --outdir fastqc/{wildcards.smp}
"""Snakefile du projet
params : paramètre à utiliser dans le shell (ici la zone d'écriture)
6- Construction pour bowtie2
rule bowtie2Build:
input: GENOME
params:
basename= "reference/reference"
output:
output1="reference/reference.1.bt2",
output2="reference/reference.2.bt2",
output3="reference/reference.3.bt2",
output4="reference/reference.4.bt2",
outputrev1="reference/reference.rev.1.bt2",
outputrev2="reference/reference.rev.2.bt2"
message: """--- Indexation of the reference genome."""
shell: "bowtie2-build {input} {params.basename}"Snakefile du projet
S'il y a plusieurs inputs, il faut préciser les noms (file ou bt2) dans le shell {input.nomImput}.
Idem pour l'output (sam et out)
7- Bowtie2
rule bowtie2:
input:
file = "samples/{smp}.fastq.gz",
bt2 = "reference/reference.rev.2.bt2"
params:
index = 'reference/reference'
output:
sam = "bowtie2/bowtie-{smp}.sam",
out = "bowtie2/bowtie-{smp}.out"
message: """--- Alignment of reads with the reference genome."""
shell: 'bowtie2 -x {params.index} -U {input.file} -S {output.sam} > {output.out}'Snakefile du projet
8- Samtools view
rule samtoolsView:
input:
"bowtie2/bowtie-{smp}.sam"
output:
"samtools/bowtie-{smp}.bam"
message: """--- Binary conversion of aligned reads."""
shell: """
samtools view -b {input} > {output}
"""9- Samtools sort et index
rule samtoolsSortIndex:
input:
"samtools/bowtie-{smp}.bam"
output:
"samtools/bowtie-{smp}.sorted.bam"
message: """--- Sorting and Indexingof aligned reads."""
shell: """
samtools sort {input} -o {output}
samtools index {output}
"""Snakefile du projet
10- Htseq count
rule htseqCount:
input:
"samtools/bowtie-{smp}.sorted.bam"
output:
"htseq/count_{smp}.txt"
params:
GFF
message: """--- Count."""
shell: """
htseq-count --stranded=no --type='gene' --idattr='ID' --order=name --format=bam {input} {params} > {output}
"""Toujours la même syntaxe
Snakefile du projet
11- Création de graphiques en R
rule graphics:
input:
"htseq/count_{smp}.txt"
output:
"graphics/graphics-{smp}.pdf"
params:
GFF
message: "--- Histogram"
shell: """
Rscript ../R-code/graphics.R {input}
"""Il est possible d'appeler toutes les commandes disponibles dans le shell comme R
Snakefile du projet
Et voilà !
Lancer un snakefile ?
snakemakeComment l'intégrer dans notre code ?
##------------------------------------------------------------------------------
## FAIR script
## Author: T. Denecker & C. Toffano-Nioche
## Affiliation: I2BC
## Aim: A workflow to process RNA-Seq.
## Organism : O. tauri
## Date: Jan 2019
## Step :
## 1- Create tree structure
## 2- Download data from SRA
## 3- Run workflow (snakemake)
##------------------------------------------------------------------------------
echo "=============================================================="
echo "Creation of tree structure"
echo "=============================================================="
mkdir Project
mkdir Project/samples
mkdir Project/annotations
mkdir Project/bowtie2
mkdir Project/fastqc
mkdir Project/genome
mkdir Project/graphics
mkdir Project/htseq
mkdir Project/reference
mkdir Project/samtools
echo "=============================================================="
echo "Download data from SRA"
echo "=============================================================="
# Get accession number (comment / uncomment to change methods)
Accession="$(cut -f7 conditions.txt | tail -n +2)" # ascp method
cd Project/samples
IFS=$'\n' # make newlines the only separator
for j in $(tail -n +2 ../../conditions.txt)
do
access=$( echo "$j" |cut -f7 )
id=$( echo "$j" |cut -f1 )
echo "--------------------------------------------------------------"
echo ${id}
echo "--------------------------------------------------------------"
ascp -QT --file-checksum=md5 --file-manifest=text --file-manifest-path=. -l 300m -P33001 -i /home/miniconda3/etc/asperaweb_id_dsa.openssh ${access} .
md5_local="$(md5sum $id.fastq.gz | cut -d' ' -f1)"
echo $md5_local
if grep -q $md5_local *.txt
then
echo "Done"
else
rm $id.fastq.gz
access=$( echo "$j" |cut -f6 )
wget ${access} # wget method
fi
rm *.txt
done
cd ../..
echo "=============================================================="
echo "Download annotations"
echo "=============================================================="
wget https://raw.githubusercontent.com/thomasdenecker/FAIR_Bioinfo/master/Data/O.tauri_annotation.gff -P Project/annotations
echo "=============================================================="
echo "Download genome"
echo "=============================================================="
wget https://raw.githubusercontent.com/thomasdenecker/FAIR_Bioinfo/master/Data/O.tauri_genome.fna -P Project/genome
echo "=============================================================="
echo "Snakemake"
echo "=============================================================="
snakemake
echo "=============================================================="
echo "Create unique count table"
echo "=============================================================="
Rscript R-code/countTable.RNouveauté : Aspera (optionnel)
Le fichier peut être corrompu ! Il faut faire des vérifications !
Ne fonctionne pas sur tous les serveurs (à coupler avec une autre méthode)
Super rapide !
Comment lancer tout le projet?
Presque comme avant!
Il faut dire au docker qu'il peut prendre de la place en CPU (ici 6) et en mémoire (ici 24 Go)
sudo docker run --rm -d -p 8888:8888 --cpus="6" -m=24g --name fair_bioinfo -v ${PWD}:/home/rstudio tdenecker/fair_bioinfosudo docker exec -it fair_bioinfo bash FAIR_script.shPuis lancer notre script
Et attendre ...
Attention pour le cloud IFB
La majorité de l'espace disque se trouve dans mnt.
Travailler dans /mnt/
Pour y aller
cd /mnt/Puis chargement du dépôt, etc
git clone https://github.com/thomasdenecker/FAIR_Bioinfo.gitFinalement, avons-nous gagné du temps ?
Machine 1
1 CPU et 2 Go de RAM
1h 23 mins
Finalement, avons-nous gagné du temps ?
Machine 1
1 CPU et 2 Go de RAM
1h 23 mins
Machine 2
8 CPUs et 32 Go de RAM
0h 28mins
-1h pour la même analyse !!
Récupération des résultats
En terminal
scp ubuntu@134.158.247.116:/mnt/FAIR_Bioinfo/Project/countTable.txt countTable.txtscp : cp (copy) en ssh
scp acces:localisationFichierDistant localisationLocal

Filezilla
Interface pour récupérer les fichiers sur le serveur
1- Renseigner la clé à Filezilla
Edition > Paramètres > SFTP
Ajouter un fichier de clé privée (le fichier que nous avons créé pour nous connecter au cloud)

./ssh/id_rsaEt pas la id_rsa.pub
Se connecter avec Filezilla
Si en ssh : ubuntu@134.158.247.116
Hôte : 134.158.247.116
Identifiant : ubuntu
Mot de passe : (rien - clés publique/privée)
Port : 22
Le message suivant sera "Clé de l'hôte inconnue"
Répondre ok


Click
Récupération des données
Architecture cliquable du serveur
Glisser / déposer
Toujours plus vite !
Cluster ifb

Un cluster ?
Regroupement de plusieurs ordinateurs indépendants (node) pour permettre une gestion globale et dépasser les limitations d'un ordinateur
Avantages
- Disponibilité augmentée (une fois le travail terminé les ressources sont libérées)
- Optimisation de la répartition du travail
- Gestion simplifiée des ressources (processeur, mémoire vive, disques durs, bande passante réseau)
Inconvénient ? 100 % terminal ?
La différence entre cloud et cluster
Cloud
Cluster
Localisation
Dispersée
Local
Matériels
Différents
Identique
Configuration
Différente
Identique
- Cloud : accumulation de puissance pour former un nuage (serveur)
- Cluster : connexion de plusieurs machines optimisées pour en former une seule
Comment lancer les calculs sur le cluster?
1. Se connecter au cluster
IFB
ssh ctoffanonioche@core.cluster.france-bioinformatique.frssh thomas.denecker@passerelle.i2bc.paris-saclay.frI2BC
2. Récupération du project sur Github
git clone https://github.com/thomasdenecker/FAIR_Bioinfo.git3. Se déplacer dans le projet
cd FAIR_Bioinfossh thomas.denecker@I2BC-Cluster.calcul.i2bc.paris-saclay.frPuis
Comment lancer les calculs sur le cluster?
4. Récupération de l'image docker dans singularity
singularity pull docker://tdenecker/fair_bioinfoSingularity
Système de conteneurs basé sur des images
Permet de fixer l'environnement
Pas besoin d'être root

Comment lancer les calculs sur le cluster?
5. Création d'un fichier pour slurm
Simple Linux Utility for Resource Management
- Allocation de ressource pour le temps nécessaire
- Suivi des tâches
- Mise en place d'une file d'attente

Le fichier slurm
Les paramètres pour le planificateur slurm commencent par #SBATCH
- fichier de redirection de l'output standard renommé avec l'identifiant du job
#SBATCH -o slurm.%N.%j.out
1- Fichier de type bash
#!/bin/bash2- Paramètres
#SBATCH -e slurm.%N.%j.err- fichier de redirection de l'erreur standard renommé avec l'identifiant du job
Le fichier slurm
2- Paramètres
#SBATCH --partition fast
- Choix des queues/nœuds de calcul
"fast" => le calcul sera coupé après n heures
#SBATCH --mem 24GB- Réservation de RAM
3- Lancement
Lancement du docker et du script FAIR_script.sh
singularity exec -B $PWD:/home/rstudio fair_bioinfo.simg bash ./FAIR_script.sh#SBATCH --cpus-per-task 6 - Nombre de CPU réservés
Le fichier slurm
Le fichier complet : fair_bioinfo.slurm
#!/bin/bash
# les parametres pour le planificateur slurm commencent par "#SBATCH" :
#SBATCH -o slurm.%N.%j.out # fichier de redirection de l'output standard renommé avec l'identifiant du job
#SBATCH -e slurm.%N.%j.err # fichier de redirection de l'erreu standard renommé avec l'identifiant du job
#SBATCH --partition fast # choix des queues/noeuds de calcul ("fast" => le calcul sera coupé après n heures)
#SBATCH --cpus-per-task 8 # Nombre de CPU réservés
#SBATCH --mem 30GB # réservation de RAM
# lancement du docker et du script FAIR_script.sh
singularity exec -B $PWD:/home/rstudio fair_bioinfo.sif bash ./FAIR_script.shsingularity v 3.0.1
Le fichier qsub
Le fichier complet : fair_bioinfo.qsub
#!/bin/sh
#PBS -q lowprio
#PBS -l select=1:ncpus=6
cd /home/thomas.denecker/FAIR_Bioinfo/
singularity exec -B /home/thomas.denecker/FAIR_Bioinfo/:/home/rstudio fair_bioinfo.simg bash ./FAIR_script.shAutre système pour mettre à la queue les jobs
Syntaxe un peu différente mais même philosophie
singularity v 2.6
Comment lancer les calculs sur le cluster?
5- Lancement du workflow
sbatch fair_bioinfo.slurm6- Récupération des fichiers (en local)
Exemple avec le cluster de l'IFB
scp ctoffanonioche@core.cluster.france-bioinformatique.fr/FAIR_Bioinfo/countTable.txt countTable.txtEt voilà !
qsub ./fair_bioinfo.qsubEt le temps de calcul?
Machine 1
1 CPU
2 Go de RAM
1h 23 mins
Machine 2
8 CPUs
32 Go de RAM
0h 28mins
Machine 2
8 CPUs
32 Go de RAM
0h 22mins
Cloud
Cluster
Conclusion
SO FAIR !
FAIR raw data
+
"FAIR_bioinfo" scripts/protocols
=
FAIR processed data
Bilan de la session
Reproductibilité et vitesse
- Mise en place d'un système automatique de workflow
- Utilisation de la puissance de parallélisation
- Augmentation des performances d'un docker
- Utilisation d'un cluster de calcul

Snakemake
Cluster
Pour la prochaine fois
Lancer l'analyse sur le cluster ou sur une VM en parallèle

Bon courage !
RDV sur Slack en cas de problème