Omiques
Qu’appelle-t-on les « omiques » ?
Origine et définition
Le suffixe -ome utilisé dans « omique » prend racine dans le sanscrit signifiant totalité. Ainsi, l’ensemble des gènes s’appellera le génome et les connaissances structurant cet ensemble constitueront la génomique Dans la même logique, l’ensemble des transcrits portera le nom de transcriptome et ses connaissances la transcriptomique
Pourquoi parlons-nous de technologies « omiques » ?
Sur la base des travaux de Jacques Monod (années 50-60), la biologie moléculaire a pour objectif de comprendre les lois de la biologie à partir des molécules constituant le vivant. Les chercheurs américains utilisent l’appellation biologie moléculaire pour désigner les techniques permettant de faire de la biologie moléculaire. Pour cette raison, il est courant de trouver dans la littérature l’utilisation de techniques omiques pour réaliser de la science omique.
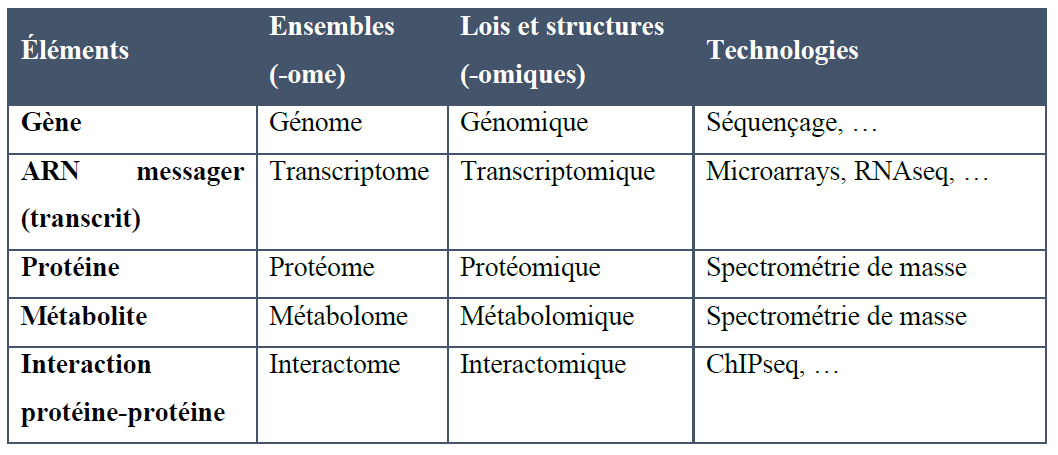
Les principales omiques et les technologies associées
Le tableau suivant regroupe les principales omiques et les technologies associées. Il est inspiré de l’article de Jacques Haiech et al (Haiech et al. 2012).
Evolution des omiques
Evolution de la génomique
Depuis les années 70, le séquençage de l’ADN repose principalement sur la méthode de Sanger (Figure 1). Cette approche impose que chaque séquence soit amplifiée puis lue individuellement ce qui en constitue sa principale limite. Malgré des améliorations considérables en matière d’automatisation et de débit, le séquençage de Sanger reste relativement coûteux et exigeant en main-d’oeuvre.
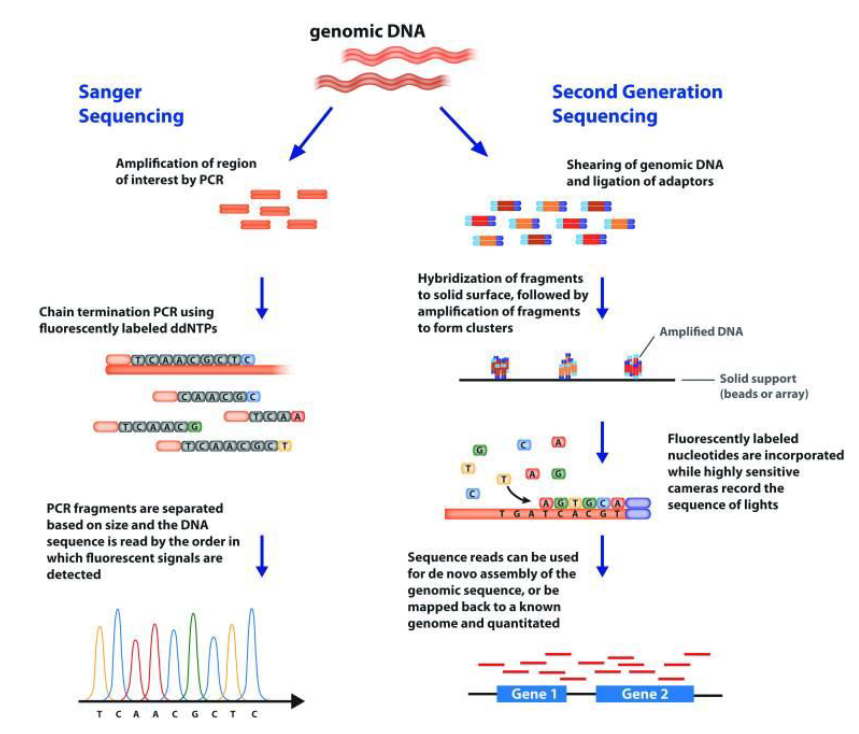
En 2005, une nouvelle génération de technologie de séquençage a vu le jour : le séquençage haut débit (NGS). Sa capacité à traiter des données en parallèle permet aujourd’hui l’analyse de plusieurs centaines de gigabases à chaque passage dans l’instrument.
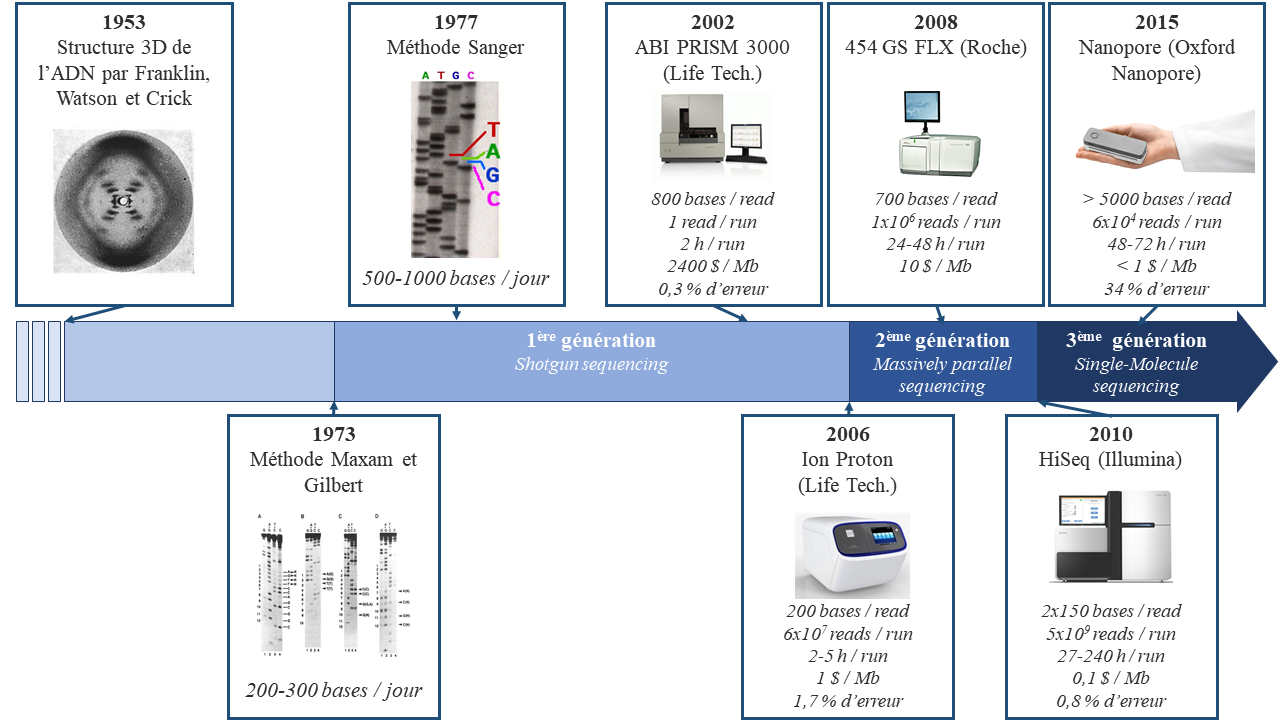
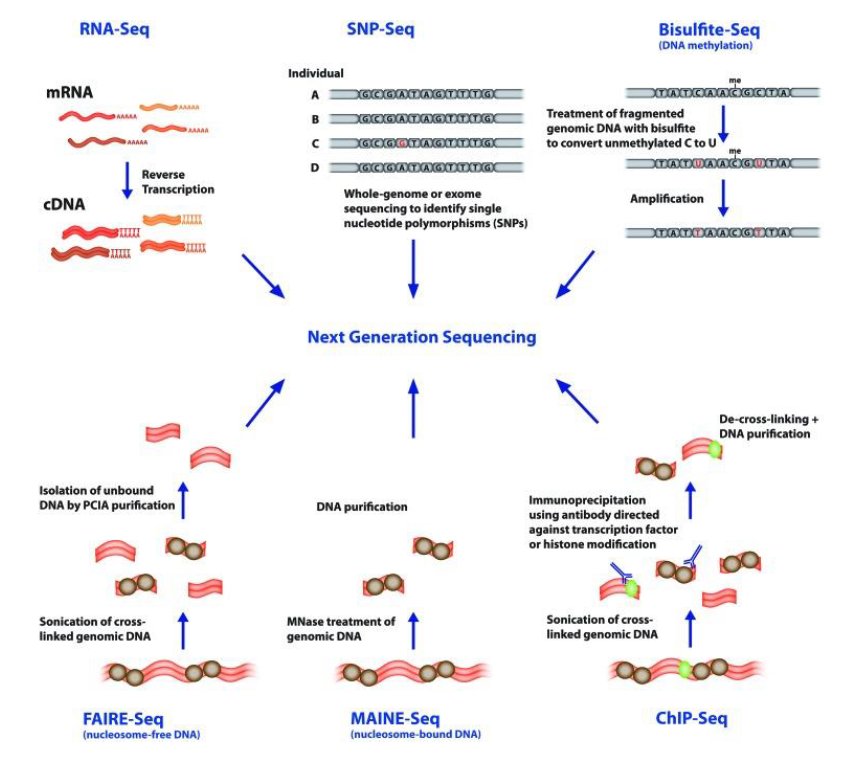
Avec les années, ces technologies sont devenues de plus en plus accessibles permettant d’élargir constamment les champs de recherche. De nouvelles techniques expérimentales ont vu le jour à l’aide de ces technologies à haut débit (Figure 3).
Evolution de la transcriptomique
A ses débuts, l’analyse des transcrits était réalisée par la technique SAGE, développée en 1995 (van Ruissen et al. 2007). Elle consiste à séquencer des « étiquettes » qui correspondent aux fragments 3′ de l’ARN messager (ARNm). Une liste d’étiquettes est ainsi obtenue et l’abondance d’un gène est proportionnelle au nombre d’étiquettes associées à ce gène. Malgré plusieurs améliorations apportées au protocole original, la technique SAGE n’est plus la technique principalement utilisée. En effet, elle nécessite beaucoup de travail et son débit est relativement faible par rapport aux applications NGS récemment développées. En parallèle, les premières puces à ADN ont été développées pour mesurer les niveaux d’expression d’un grand nombre de gènes (Schena et al. 1995). Les transcrits isolés d’un échantillon d’intérêt sont (1) convertis en ADNc ou ARNc, (2) marqués puis (3) hybridés à la puce à ADN. La quantité d’hybridation détectée pour une sonde spécifique est proportionnelle au niveau de transcription du gène correspondant. La comparaison des niveaux de transcription entre divers types de cellules ou conditions peut être utilisée pour identifier les gènes qui sont impliqués dans la différenciation des cellules ou dans les réponses à certains changements environnementaux. Les puces à ADN sont toujours utilisées aujourd’hui même si elles sont peu à peu remplacées par des techniques basées sur le NGS comme le RNAseq. Le RNAseq consiste à lire des séquences dérivées d’un échantillon d’ARNs d’intérêt. Ces séquences d’ARN sont comparées avec un génome de référence pour les associer à un gène. Le nombre de séquences d’ARN associées à un gène représente le niveau d’expression de ce gène. Le RNAseq est très rapide et propose une plus grande couverture du génome mais représente un coût économique important par rapport au puce à ADN.
Evolution de la protéomique
L’évolution de la protéomique a lieu à plusieurs niveaux :
- La séparation des protéines – Initialement les protéines étaient séparées par gel bidimensionnel par taille puis par charge. Cette approche a été remplacée par une séparation par chromatographie liquide ;
- A partir de protéines fragmentées ou non fragmentées ? Deux stratégies d’identification s’opposent actuellement. La différence repose sur la fragmentation ou non de la protéine en peptides. Même si l’identification des protéines non fragmentées est plus fiable (top down), les défis techniques qu’elle entraine conduisent à privilégier actuellement l’identification par fragmentation (bottom up) ;
- Les techniques de spectrométrie de masse – Il existe de nombreuses techniques pour déterminer le poids moléculaire d’un peptide comme par mesure du « temps de vol » (TOF), par trappe ionique, etc