Spectrométrie de masse
Principes et composants
Un peu d’histoire
La spectrométrie de masse est une technique d’analyse physico-chimique permettant de détecter, d’identifier et de quantifier des molécules d’intérêt par mesure de leur masse moléculaire exacte, rapportant ainsi à leur composition élémentaire. Elle peut être vue comme une balance moléculaire. Cette technologie prend racine en 1897 lorsque J. Thomson découvre l’électron et détermine son rapport m/z (Figure 1). Le premier spectromètre de masse est construit 15 ans plus tard en 1912. Depuis plus de 100 ans, nous utilisons cette technologie qui ne cesse d’être améliorée (sensibilité, précision, etc). L’apparition de nouvelles sources d’ionisation dans les années 80 ont grandement amélioré le domaine : le Maldi par K. Tanaka (1987) et l’ESI par J.B. Fenn (1988). Ils ont tous les deux étés récompensés pour leurs travaux par le prix Nobel de Chimie en 2002. Elle est utilisée dans de nombreux domaines comme la physique, la biologie, la médecine1.
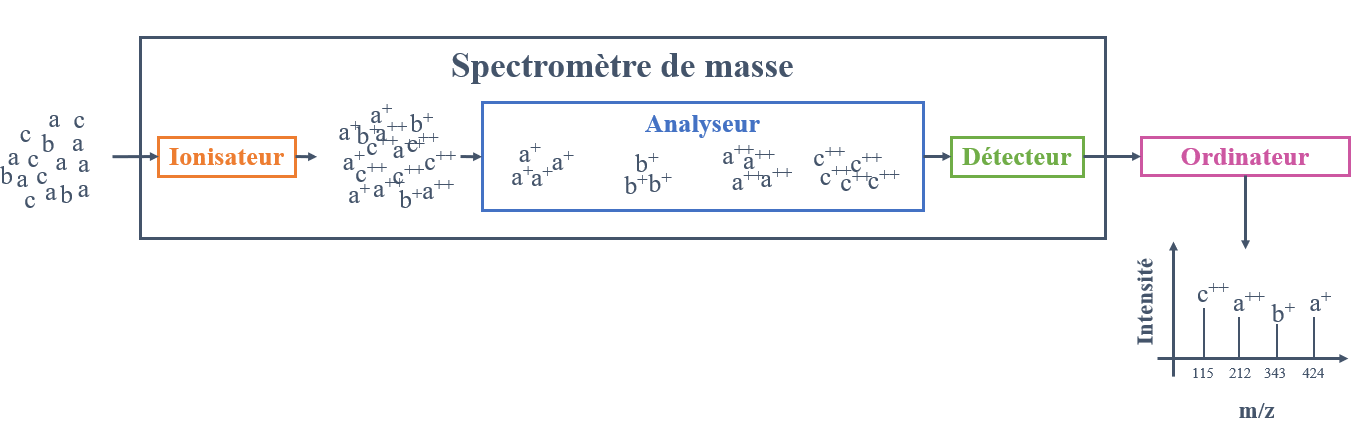
Composition d’un spectromètre de masse
Le principe de la spectrométrie de masse est d’analyser et déterminer la masse exacte d’une molécule chargée en fonction de son rapport masse/charge (m/z). Un spectromètre de masse est composé de 4 grands éléments :
- Un ionisateur (ou aussi de source d’ionisation) – Il volatilise et ionise les éléments présents dans un échantillon. Les ions sont produits en phase gazeuse à partir des états solides, liquides ou gazeux ;
- Un ou plusieurs analyseurs – Il permet la distinguer des éléments chargés en fonction de leur rapport masse sur charge (m/z) ;
- Un détecteur – Le détecteur va ensuite « compter » les différents ions et amplifier leurs signaux. Il convertira le courant ionique en courant électrique. Le signal obtenu sera proportionnel à la quantité d’ions reçus. Ces ions sont détectés soit à la suite d’une oscillation dont la fréquence est spécifique à leur rapport m/z (technologie Orbitrap) soit à la suite du calcul du temps de vol de l’ion qui est spécifique du rapport m/z (principe du TOF) ;
- Un ordinateur – Il collecte et traite les signaux puis génère un spectre de masse.
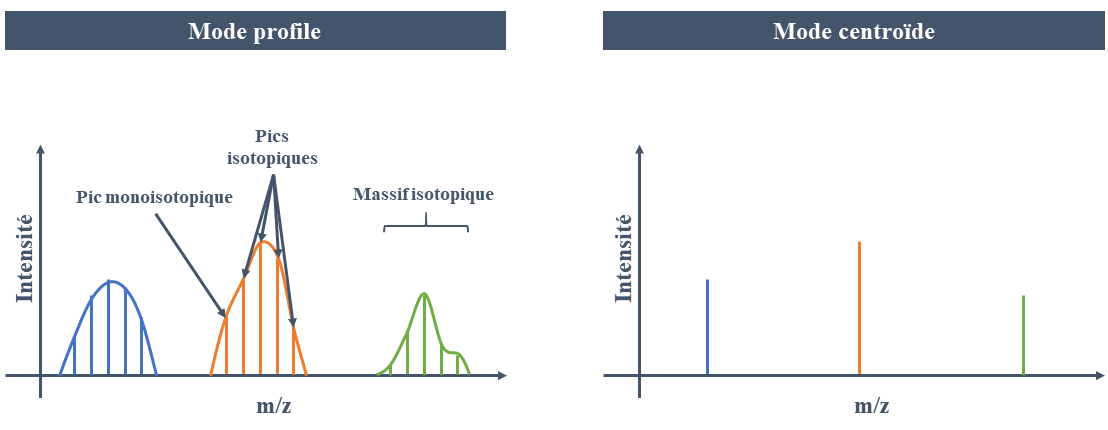
Spectres de masse
Le spectre de masse est un graphique représentant l’intensité en fonction du rapport masse sur charge (m/z). La masse est exprimé en dalton (1 Da = 1,673.10-27 kg) et le rapport m/z en Thomson (Th). Les spectres de masses peuvent être stockés de deux façons : soit en conservant toutes les données, soit en traitant les données pour les alléger.
Lorsque toutes les données sont conservées, nous parlons de spectres de données brutes (ou spectres continus ou spectres en mode profil). Les données brutes sont celles les moins retraitées et les plus proches de celles du détecteur. Un élément ionisé détecté est représenté dans le spectre comme un pic dont la hauteur correspond à son intensité et la position son rapport masse sur charge (m/z). Pour un élément correspondant à un m/z donné, comme un peptide, il est possible d’observer plusieurs pics m/z. Donc ce cas, on parle de massifs isotopiques, c’est-à-dire la combinaison de plusieurs isotopes. L’écart entre les pics d’un même massif isotopique est équivalent et permet de rapporter la charge du peptide analysé.
Chacun des pics du massif sont le même élément mais dont la composition est différente (présence d’isotope, voir encadré ci-dessous). Le premier pic du massif est le pic monoisotopique. Il n’est composé que par les isotopes les plus abondants (C12, H1, O16…). La masse de ce pic est la masse monoisotopique. Il s’agit de la masse exacte de l’élément. Les autres pics du massif isotopique sont appelés les pics P+1, P+2, … Ils contiennent tous au moins 1 des isotopes lourds de l’éléments. Pour obtenir la masse de l’élément la plus précise, il faut multiplier cette masse monoisotopique par la charge du massif isotopique2. Les fichiers contenant ces données ont comme extension .raw. La Figure 3 (gauche) présente un exemple de spectre de données brutes. Dans cette étude, nous n’avons travaillé qu’avec ce type de fichiers. La grosse limite est le volume de stockage (plusieurs gigaoctets) qui complique énormément leur traitement.
- Son symbole chimique M (comme H pour l’Hydrogène, C pour le Carbone, etc) ;
- Son nombre de masse A (égal au nombre de nucléons de l'atome), placé en haut et à gauche du symbole chimique ; A = N + Z
- Son numéro atomique Z, placé en bas et à gauche du symbole chimique.
Pour alléger les fichiers, les spectres peuvent être transformés en liste de pics où toutes les mesures pour chaque massif isotopique sont combinées en une seule valeur de masse. Nous parlons dans ce cas de spectre en mode centroïde. La valeur associée à chaque massif isotopique est généralement celle de l’élément isotopique présentant l’intensité maximale. Les fichiers contenant ces informations sont par conséquent plus légers et plus simples à manipuler mais de l’information a été perdue. Ces fichiers ont généralement comme extension le mzml. Un exemple est proposé Figure 3 (droite).
Spectrométrie de masse en tandem LC-MS/MS
Appareillage
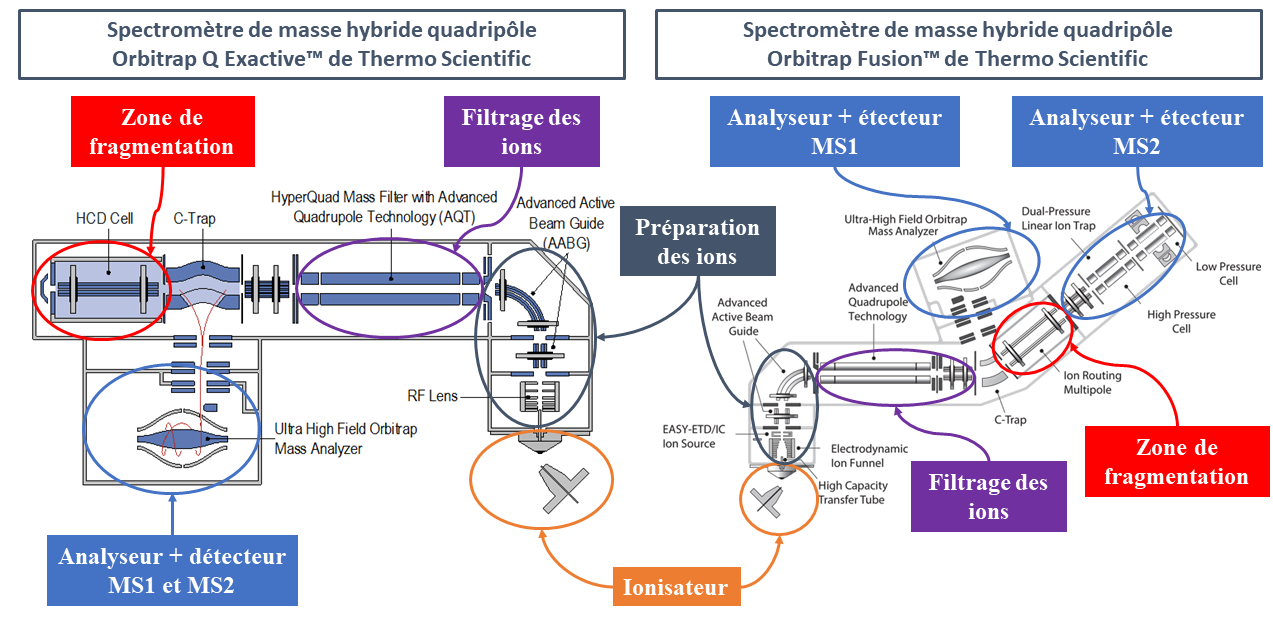
Pour identifier des protéines, actuellement, l’approche la plus courante est la spectrométrie de masse en tandem LC-MS/MS par stratégie ascendante Bottum up. Sur la plateforme de protéomique de l’Institut Jacques Monod, nous pouvons retrouver deux types d’appareils : le spectromètre de masse hybride quadripôle Orbitrap Q Exactive™ plus et le spectromètre de masse Tribrid™ Orbitrap Fusion™ de Thermo Scientific™. Ils fonctionnent exactement sur le même principe. La seule différence est que le Fusion™ est composé de deux analyseurs/détecteurs permettant de paralléliser les analyses. Sur la Figure 4, l’ensemble des composants présentés dans le paragraphe « Composition d’un spectromètre de masse » (ci-dessus) sont mis en avant sur les schémas techniques des deux appareils.
Différentes étapes
Les différentes étapes d’une analyse par spectrométrie de masse en tandem LC-MS/MS en approche Bottom up sont détaillées ci-dessous :
1. Extraction protéique
La première étape consiste à extraire les protéines de l’échantillon. Cette étape est particulièrement délicate et critique puisqu’il faut trouver l’équilibre entre d’une part récupérer le maximum de protéines dans le plus de compartiments possibles et d’autre part limiter un maximum la dégradation des protéines.
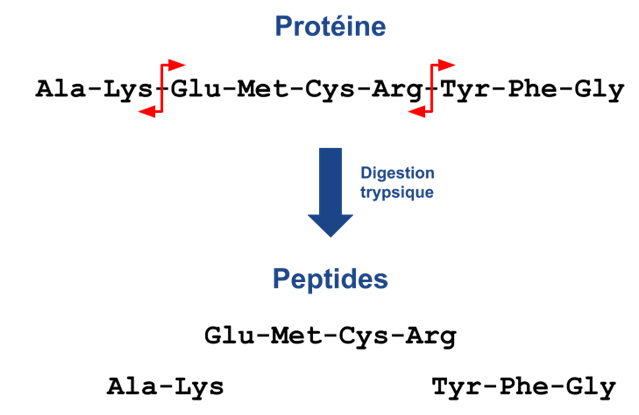
2. Digestion trypsique
Une fois que l’ensemble des protéines sont récupérées, l’étape suivante consistera à les digérer à l’aide d’une enzyme (trypsine) pour obtenir un mélange de peptides (approche Bottom up). La trypsine est utilisée pour réaliser cette digestion dont une visualisation 3D est Figure 5 - A (trypsine humaine). Il s’agit d’une endoprotéase3 qui peut être vue comme des ciseaux moléculaires coupant à des endroits très précis. Le site d’hydrolyse pour la trypsine est le groupement carboxyle des acides aminés lysine et arginine (sauf si elles sont suivies d’une proline). Un exemple d’une digestion trypsique est présenté à la Figure 5-B.
3. Séparation des peptides
Une fois la digestion terminée, l’objectif à présent sera de séparer les différents peptides composant le mélange en fonction de leur hydrophobicité. Pour les séparer, nous utilisons la chromatographie en phase liquide (liquid chromatography ou LC en anglais). Cette technique entraine les peptides dans un liquide (phase mobile) à travers une phase solide stationnaire. En fonction des propriétés physico chimiques des peptides, ils seront relargués plus ou moins rapidement. Dans ce type de chromatographie liquide en phase inverse, un peptide peu hydrophobe sortira beaucoup plus rapidement qu’un peptide hydrophobe. En LC-MS/MS (Liquid Chromatography - Mass Spectrometry / Mass Spectrometry), les types de chromatographie utilisées sont généralement de type HPLC (High Performance Liquid Chromatography) voir UHPLC (Ultra High Performance Liquid Chromatography) pour obtenir une séparation la plus fine possible. Les colonnes utilisées sont de type C18 (chromatographie en phase inverse ou reverse phase). Au laboratoire, l’appareil utilisé pour réaliser la chromatographie est une Thermo Scientific™ EASY-nLC™ Proxeon 1200.
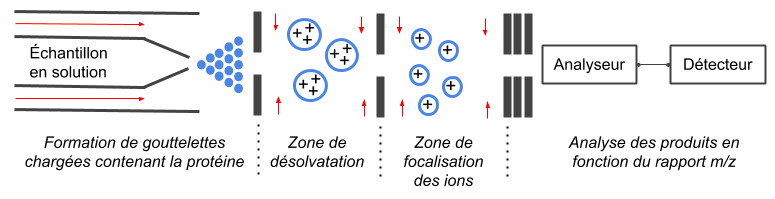
4. Ionisation des peptides
Une fois les peptides séparés, ils sont injectés dans le spectromètre de masse pour y être ionisés. Au laboratoire, nous utilisons la technique d’électronébulisation (ou electrospray) très fréquente après une chromatographie en phase liquide. Les peptides en phase liquide vont être chargés et dispersés sous forme de gouttelettes. Un gaz va ensuite être appliqué sur ces gouttelettes pour éliminer le liquide (désolvatation). Lorsque la limite de Raleigh est atteinte, les forces de répulsion coulombienne entre les charges vont être tellement fortes qu’elles vont engendrer une « explosion Coulombienne » pour former des gouttelettes plus petites (Wilm 2011). Ce processus est répété jusqu’à obtenir des gouttelettes ne contenant qu’un peptide chargé. Les peptides vont ensuite passer par un ensemble de zones pour les focaliser (Slens) et éliminer les éléments non chargés (Active Beam Guide).
5. Filtrage
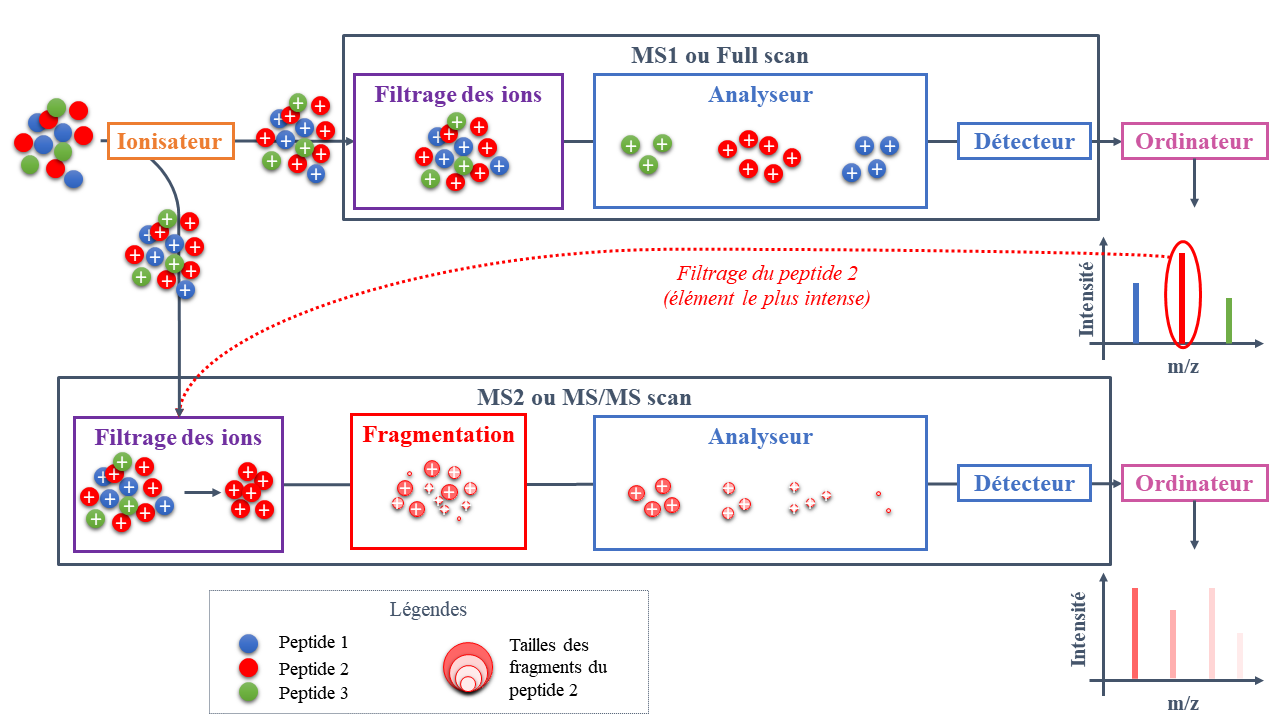
Une fois les éléments prêts à être analysés, ils sont envoyés dans le quadripôle utilisé pour le filtrage des éléments. En MS1, aucun filtrage n’est réalisé : tous les peptides chargés passent.
En MS1, les éléments ionisés sont des peptides.
6. Analyseur
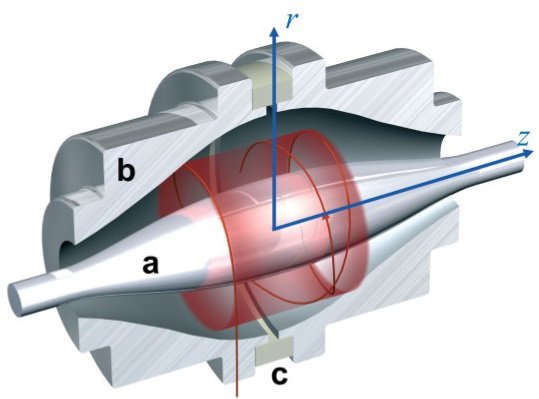
Il s’agit du premier passage dans un analyseur. L’objectif est de différentier les peptides ionisés collectés. La façon de séparer les éléments ionisés est dépendante du type d’analyseur utilisé4. Au laboratoire, la technologie utilisée est l’Orbitrap de Thermo Fisher. Le principe est de piéger les ions par un champ électrostatique. Ces ions sont séparés grâce à leur différence de fréquence d’oscillation axiale (dépendante de m/z). L’Orbitrap est composée de deux électrodes : une centrale qui piège les ions dans un mouvement orbitale et une externe en forme de cylindre ainsi qu’une électrode externe en forme de cylindre (Figure 8).
7. Détecteur
Une fois séparés, les peptides ionisés vont être détectés grâce à leur mouvement. Il convertira le courant ionique en courant électrique. Le signal obtenu sera proportionnel à la quantité d’ions reçus.
8. Ordinateur
Il collecte et traite les signaux puis génère un spectre de masse. Chaque pic correspondra à un peptide. Informatiquement, les peptides les plus intenses (les pics les plus haut) à une charge donnée seront sélectionnés pour être fragmentés puis identifiés. Sur la plateforme, les 20 peptides les plus intenses à une charge donnée vont être fragmentés (mode DDA5). Sur la Figure 7, pour simplifier la compréhension, un seul peptide a été filtré (le peptide 2 en rouge).
En MS2, chaque peptide sélectionné va subir le même traitement. Pour simplifier la compréhension, la suite sera décrite pour un seul peptide.
9. Filtrage
Le quadripôle va cette fois ci jouer son rôle de filtrage. Il ne va conserver que le peptide sélectionné à une charge donnée. Seul ce peptide à une charge donnée va passer à l’étape suivante.
10. Fragmentation
La fragmentation permet d’obtenir un niveau d’information supplémentaire : la structure. Dans ce cas, pour les peptides, il s’agit de la séquence en acides aminés dont l’étape MS1 donne la masse précise. Le peptide va être fragmenté en petits fragments de séquence dans une cellule de collision. La fragmentation du peptide se fait principalement le long de son squelette carboné. Si la charge est portée sur la partie N-terminale, le fragment est classé dans le groupe a, b ou c en fonction du lieu de coupure (Figure 9). Si la charge est portée sur la partie C-terminale, le fragment est classé dans le groupe x, y ou z en fonction du lieu de coupure. Un nombre est ensuite associé pour indiquer le nombre de résidus associés. Chaque fragment sera donc composé de plus ou moins d’acides aminés. Cette nomenclature est utilisée sur la Figure 10.
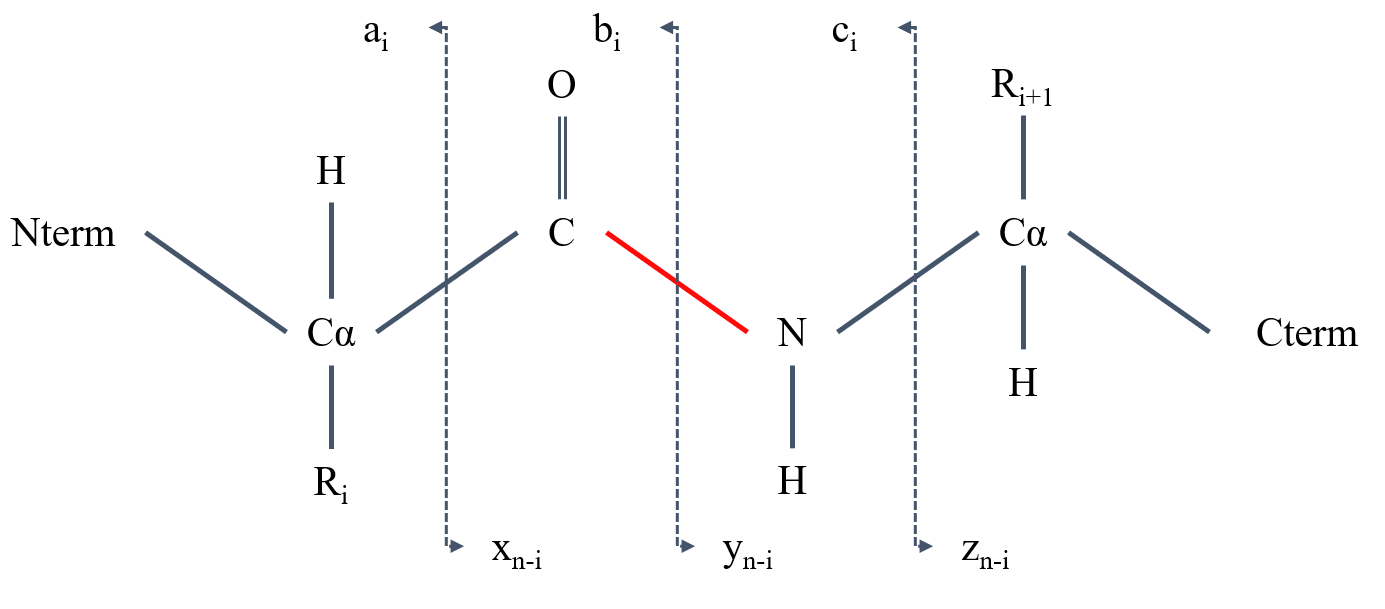
11. Analyseur
Lorsque qu’un peptide est fragmenté, nous obtenons des petits fragments de taille (et donc de masse) différentes. Cette étape va permettre de séparer par la masse les différents fragments de peptides.
12. Détecteur
Chaque pic correspondra à un fragment plus ou moins lourds en fonction de sa composition propre en acides aminés. La différence de poids entre deux pics successifs correspond exactement au poids d’un acide aminé. Ainsi, en calculant les différences de poids successivement, il est possible de déterminer la séquence du peptide qui a été fragmenté (Figure 10). À partir de cette information et du spectre de masse, il sera possible d’identifier la protéine d’origine.
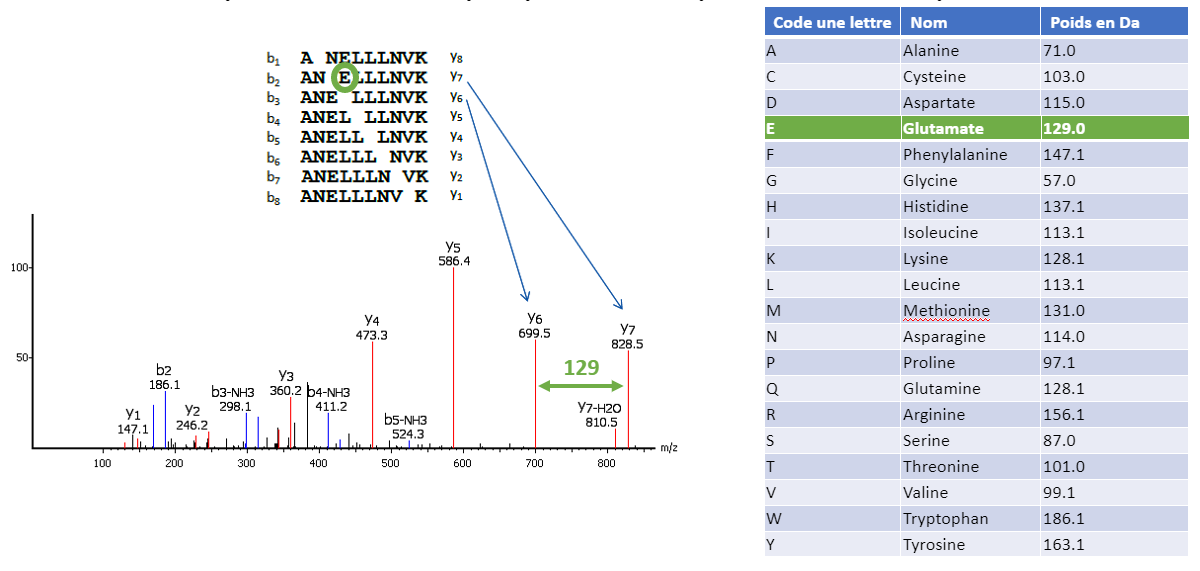
À partir de toutes les données générées, l’analyse computationnelle va permettre d’identifier les protéines d’où proviennent les peptides.
Identification des protéines à partir des peptides
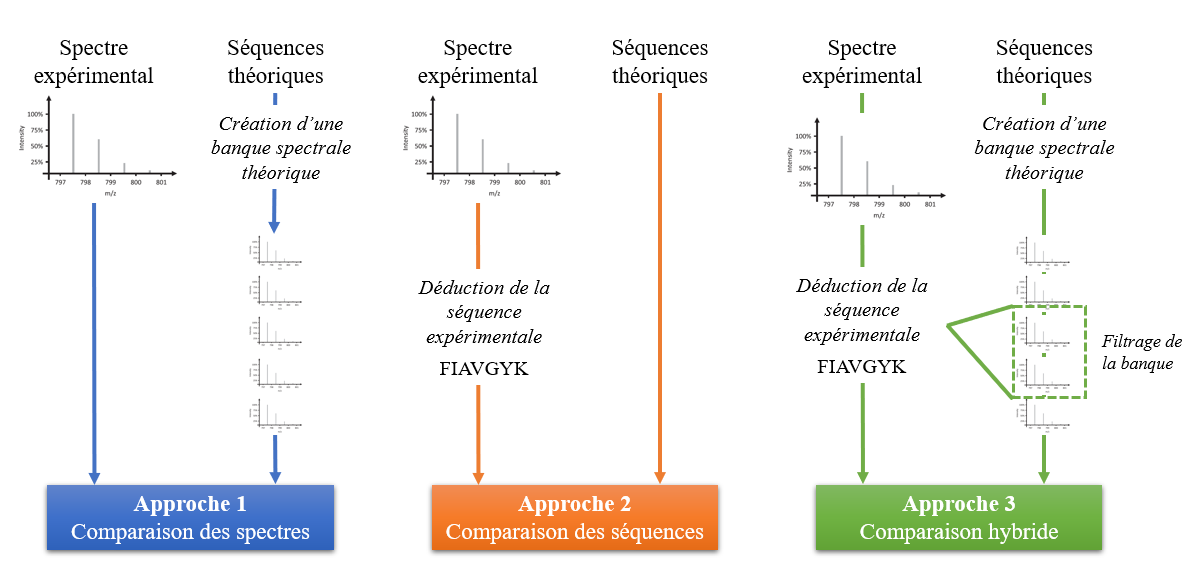
Il existe de nombreuses méthodes d’identification disponibles dont une liste très complète est disponible sur Wikipédia. Ces moteurs de recherche peuvent se séparer en 3 groupes en fonction de la stratégie utilisée (Figure 11) :
- Une recherche basée uniquement sur des banques de données – Le principe est de générer des spectres théoriques à partir des séquences des protéines (en fonction de l’enzyme de clivage, la présence possible de modifications post-traductionnelles, etc) et de les comparer aux spectres expérimentaux (ceux obtenu en MS2). Cette approche est très performante (nombreuses identifications de peptides) mais peut être très lente. Nous pouvons citer les outils Mascot (Koenig et al. 2008), Sequest (Eng et al. 1994) ou encore X!Tandem .
- Une recherche de novo indépendante des banques de données – Le principe est de déterminer la séquence peptidique à partir des spectres MS/MS (comme nous l’avons vu précédemment et illustré Figure 10) et de rechercher à quelle protéine elle est associée (par alignement de séquence). Nous pouvons citer l’outil Peaks. Généralement l’identification est plus rapide mais moins performante.
- Une recherche hybride combinant les deux approches – Le principe est de déterminer la séquence peptidique à partir de ce spectre pour (1) réaliser une sous-sélection de spectres dans une banques de données de spectres théoriques puis (2) de comparer le spectre expérimental avec ces spectres sélectionnés. Cette méthode permet plus d’identification qu’en ne faisait que la méthode de novo et est plus rapide que l’approche par banque de données puisqu’il y a une sous sélection des spectres théoriques à comparer. Nous pouvons citer l’outil Byonic (Bern et al. 2012).
Une fois l’identification réalisée, il faut lui donner un score pour évaluer sa qualité. Le score dépend de l’approche utilisée mais certains critères sont identiques :
- La différence de masse entre la masse du précurseur et la masse du peptide théorique ;
- La différence de masse entre les masses des fragments et les masses théoriques des fragments ;
- L’intensité des pics identifiés pour être un fragment et les non identifiés ;
- La corrélation de l’intensité du fragment et les intensités théoriques attendues calculés à partir des peptides théoriques.
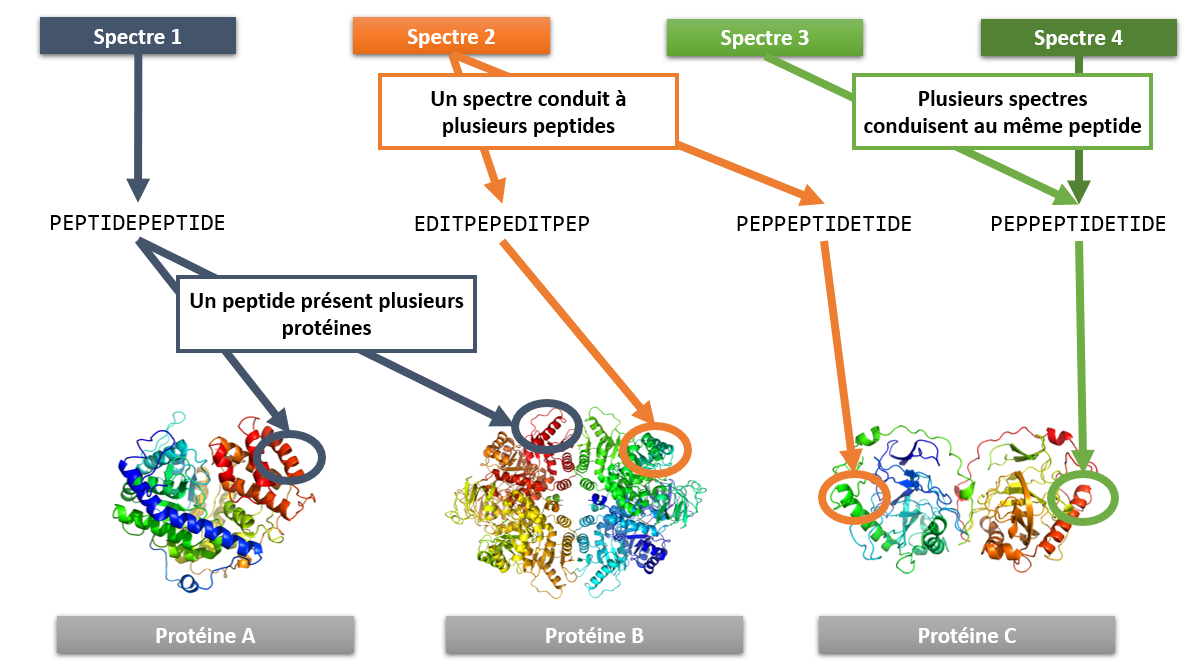
Ces critères vont permettre de calculer un score de qualité, comme une valeur P (P-value) ou une valeur E (E-value), qui seront une bonne aide pour sélectionner les identifications pertinentes. Certaines causes courantes compliquent néanmoins l’identification d’une protéine (Figure 12) :
- Un peptide identifié à partir d’un spectre peut apparaitre dans différentes protéines (peptide partagé ou commun). En effet, plusieurs protéines peuvent avoir une partie de séquence en acides aminés similaire qui donneront les peptides communs mais posséder un ou plusieurs peptides uniques permettant l’identification de la protéine de façon certaine. Dans le cas où il n’y a pas de peptide unique identifié, la protéine sélectionnée sera celle qui présentera le meilleur alignement avec le peptide. Si aucune distinction n’est possible (score identique), une liste de protéines est proposée avec comme classement en première position une protéine choisie aléatoirement, pouvant générer des faux positifs / erreurs ;
- Différents peptides peuvent être identifiés à partir du même spectre ;
- Un peptide peut être identifié à partir de plusieurs spectres.
-
J’ai personnellement utilisé plusieurs fois un spectromètre de masse pour identifier des bactéries isolées chez des patients à l’hôpital. ↩
-
Le site suivant propose une méthode détaillée pour retrouver la charge d’un massif isotopique : ici [Accessible le 22/05/2020] ↩
-
Une endoprotéase est une peptidase capable de couper à l’intérieur d’une chaine protéique. Une peptidase est une enzyme qui lyse des peptides. Enfin une enzyme est une molécule ou ensemble de molécules qui catalyse des réactions chimiques biologiques, donnant un ou des produits à partir d’un ou de plusieurs substrats. (Wikitionnaire) ↩
-
Pour en savoir plus entre les différents analyseurs, Lennart Martens propose [un cours disponible en ligne] (https://www.youtube.com/watch?v=NKXhyjsgT1I) [Accessible le 28/05/2020] ↩
-
DDA pour data-dependent acquisition. Ce mode s’oppose au mode DIA (data-independent acquisition) qui analyse tous les éléments dans des fenêtres successives. Pour en savoir plus, l’article de Hu et al propose une comparaison des deux approches, notamment sur la figure 2 (A. Hu et al. 2016). ↩