bPeaks explorer
bPeaks exmplorer allows the exploration of bPeaks analyzer results.The page is divided into 5 parts :
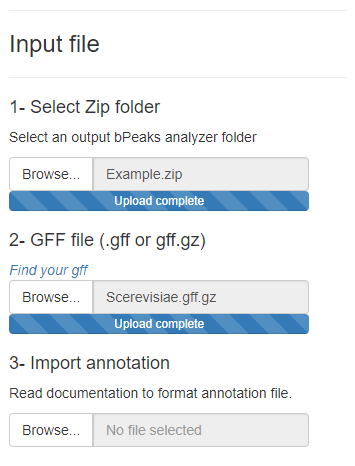
Import area
3 files can be imported:- a zipped file obtained with Bpeask (Data folder)
- an annotation file in gff or gff.gz format. It is possible to find this type of file on NCBI genome. An example file is available on Github (Data folder)
- An annotation file to fill the database. This file is composed of 8 columns: the feature name, the database name, the standard name, the beginning and end position of the gene, the chromosome, a description and the organism. The file is in TSV format (separator : tabulation). A example is available in Github (Database folder)
Summary area
The summary area is divided into 4 parts:- A bar plot representing the number of peaks detected per chromosome
- A chromosome selector to change the chromosome in the explorer
- A pdf from bPeaks summarizing the analysis results for the selected chromosome
- A table summarizing the parameters used in the analysis.

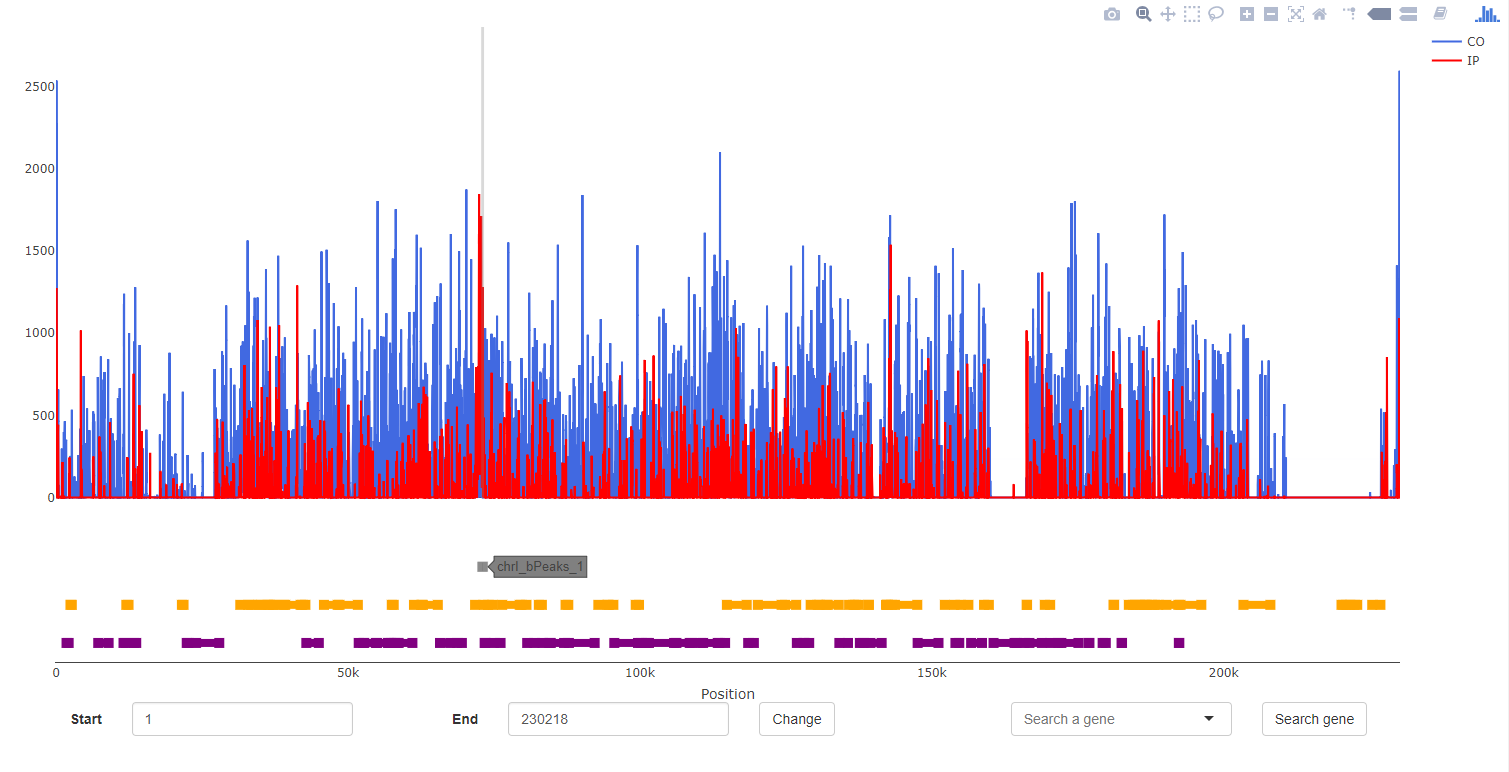
Genome viewer
The genome viewer offers a dynamic exploration of the results. It is composeb by two graphs :- Number of read mapped according to genomic position
- Annotations of peaks (gray) and genes (orange for strand + and purple for strand -). Click on extremity
Supplementary informations
Two types of additional information can be displayed in this field:
- if you click on an extremity of gene (orange or purple segment) (if a gff is imported) :
- The informations present for this gene in the database
- The informations present for this gene in the gff
- if you click on an extremity of peak (gray segment) :
- A set of information about the peak such as its position, chromosome and sequence if a fasta file with the reference genome was used in the analysis.
- A pdf with for this peak a graph of T1, T2 T3 and T4

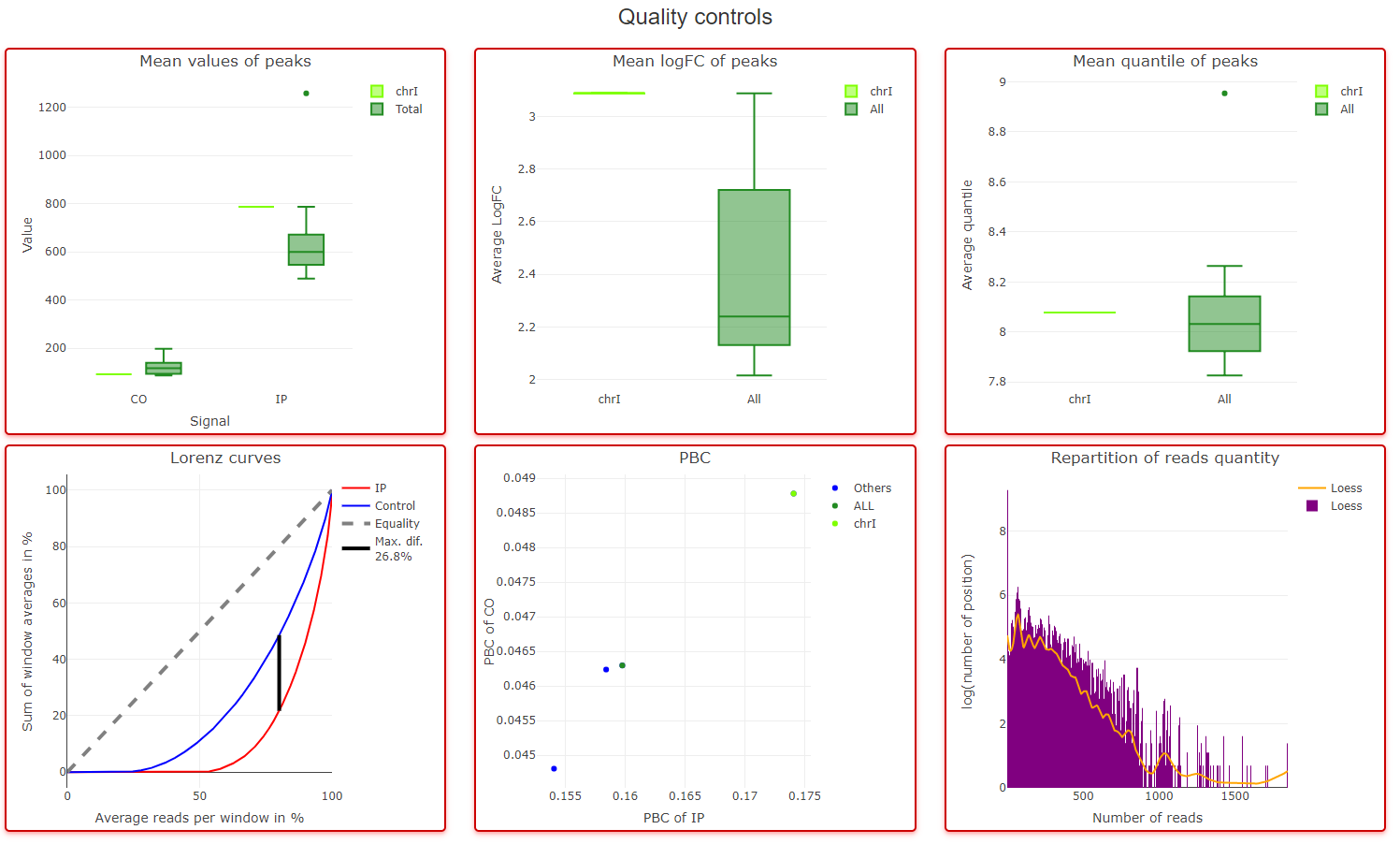
Quality control area
For each chromosome, you will find in this zone 6 graphs :
- A average number of reads in peak detected in comparison with all chromosomes (boxplots)
- A average of logFC in comparison with all chromosomes (boxplots)
- A average of quantile in comparison with all chromosomes (boxplots)
- Lorenz curves
- IP sample PBC according to Ip sample PBC
- Number of positions (logged) with a given number of reads