Why bPeaks application?
Detect protein binding sites from ChIP-seq data in small eukaryotic genomes

Peak calling is a critical step in ChIPseq data analysis. Choosing the correct algorithm as well as optimized parameters for a specific biological system is an essential task. bPeaks (Merhej J. et al.) uses simple parameters to compare the sequences (reads) obtained from the immunoprecipitation (IP) with those from the control DNA (input). Because yeasts have small genomes ( <50 Mb), our program has the advantage of using ChIPseq information at the single nucleotide level and can explore, in a reasonable computational time, results obtained with different sets of parameter values.
Running a peak caller has never been easier

Simplified file import
With a browser to select your files, a parameterization to read files and a pre-visualization of your data, importing data has become child's play.
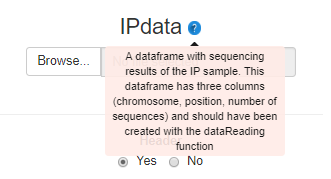
Assisted parameter setting
Each bPeaks parameter has a helper and a default value.
Data quality control
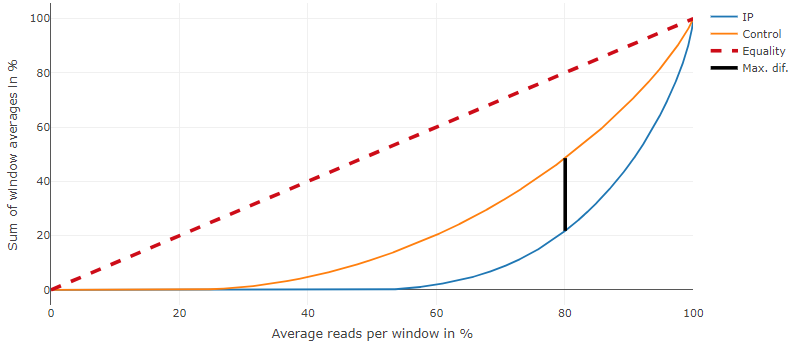
Lorenz curves
Analyze reads among the genome windows by computing cumulative sum. A bad ChIp have a straight line and a good ChIP a lorenz curve

PBC
An approximate measure of library complexity
Genome browser to explore results
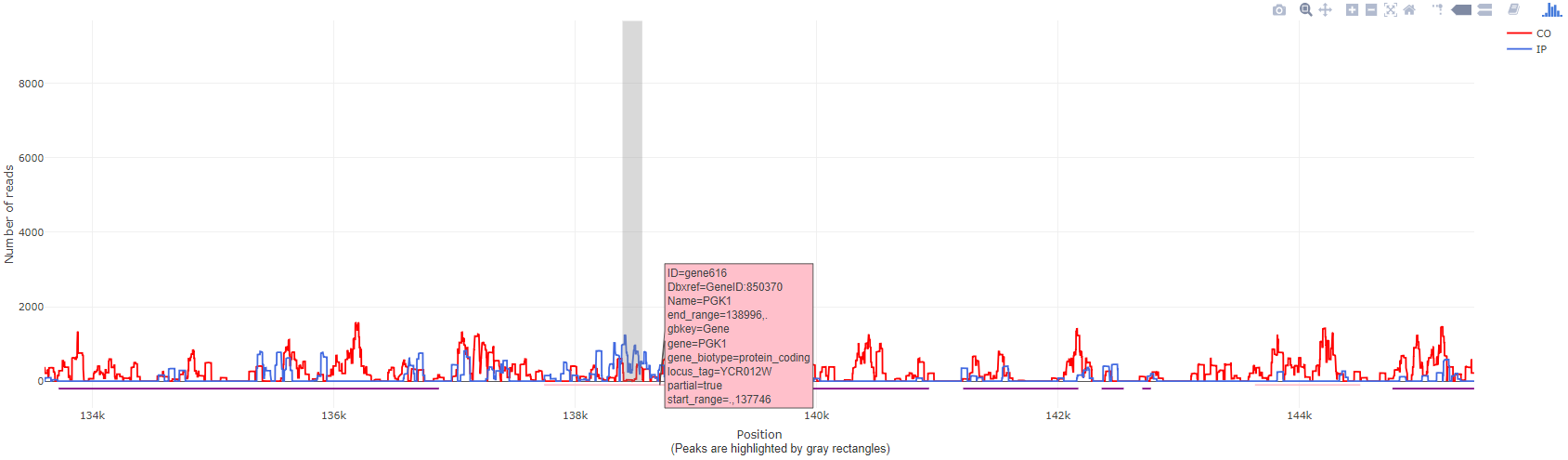
With our genome browser, the exploration of bPeaks results is very fast. It is possible to import a GFF file to obtain the annotations of the species studied.
Open source project


So, convinced?
Download it now!
bPeaks application : An intuitive peak-calling strategy to detect and visualize protein binding sites from ChIP-seq data in small eukaryotic genomes.
Download